
人染色体
第二十二章
22号染色体长51.305Mb,编码499个基因(信息参考:Ensembl GRCh37,release 109 - Feb 2023 © EMBL-EBI),基因密度约为9.73,属G组,近端着丝粒染色体(见图22-1)。相比长48.130Mb,编码243个基因的第21号染色体,22既大于它,编码的基因也比它多一倍还有余。在最初观察到Down患儿有一额外的小染色体时,第一届国际人类细胞遗传学会还未举行,肉眼的分辨能力也不够精确,故就将它排在了第21位,因为21三体是第一个被认识的染色体病,已广为大众接受,所以在发现21号染色体比22号染色体小后,也没有更改它们的位子。染色体编码的基因多,意味着这条染色体对个体生存更重要,22号染色体比13,18号染色体编码的基因都多(13号染色体编码329个基因,18号染色体编码289个基因),所以22号三体胚胎不能存活到出生,因此我们从没见过22三体患儿。

22q11.23 的BCR断裂易位形成的小染色体,就是著名的费城染色体(见图22-2)右边箭头所指,它是世界上唯一一个以城市名字命名的染色体,在普通光学显微镜下几乎就是一个小黑点!

9q34的abl癌基因与22q11的bcr基因融合形成abl/bcr融和基因,产生了一种210千道尔顿的异常蛋白质(P210),该异常蛋白表达的酪氨酸激酶活性特别高,这种高活性酪氨酸激酶在Ph+急性淋巴细胞白血病(Ph+ ALL)患者的白血病细胞中也可见。同样是t(9;22)(q34;q11),但在染色体断裂区域剪接位点却不同,Ph+ ALL形成的abl/bcr融和基因,产生的是一种190千道尔顿的异常蛋白质(P190),但是格列卫对Ph+ ALL患者仍然有效!
22q11.2的缺失与一系列发育缺陷(特别是DiGeorge综合征、velocardiofacial综合征、圆锥动脉干异常面部综合征和孤立的圆锥动脉干心脏缺陷)相关,这些缺陷被归类为首字母缩写CATCH 22。1965年美国医生Angelo DiGeorge首先描述并报道了该疾病的临床特征。因患者常伴发心脏、腭部和颜面畸形,又俗称“腭心面综合征”。在新生儿中的发病率约为1/1000-4000,无性别差异。
DiGeorge综合征又称先天性无胸腺或发育不全,属于原发性细胞免疫缺陷病。由于胚胎期第三、第四咽囊发育障碍,使胸腺和甲状旁腺缺如或发育不全而引起先天性异常。患儿常伴其它先天性畸形。由于胎儿甲状旁腺机能减退和低血钙,新生儿出现手足搐搦症,低血钙症倾向于生后1年内缓解。患儿可表现出特殊面容,如唇腭裂、耳廓畸形、眼裂下斜、眼距宽、斜视、鼻畸形、上下颌骨发育不良、面部不对称。常存在大血管异常,如法洛四联症和主动脉弓右位。如新生儿期未死亡,生后3~4个月可发生各种严重的病毒、真菌如念珠菌和卡氏肺囊虫感染。
22q11.2微缺失综合征的诊断一方面依据上述临床表现,如胸腺缺如、甲状旁腺及T细胞功能异常等,特别是几个主要表现的组合,确诊可依靠细胞分子遗传学检测。常用的实验室检测技术包括荧光原位杂交(fluorescence in situ hybridization,FISH)技术;特异性22q11.2探针TUPLE1(见图22-3红色信号,绿色信号为22号染色体上的DNA片段,作为参照),主要检测缺失。

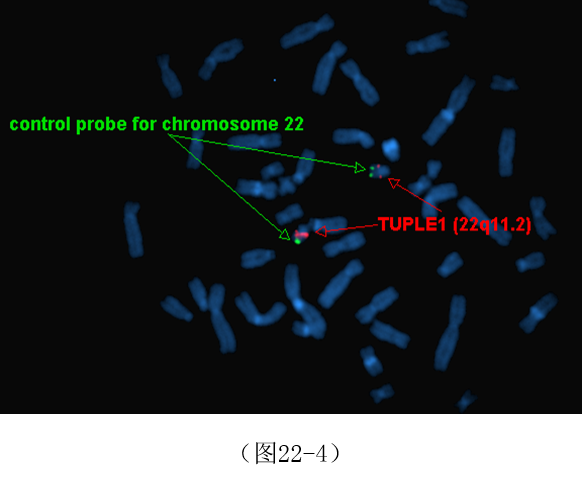
与相对常见的22q11.2缺失相比, 22q11.2的重复更罕见,并导致一系列明显的畸形和出生缺陷,称为22q11.2重复综合征(22q11.2 microduplicationsyndrome,MIM # 608363)。重复片段在1.5Mb-6.0Mb,涉及到TUPLE1、BCR、IGLL 1及RGL4等20余个基因。对这种重复的诊断通常需要通过荧光原位分析间期细胞杂交或染色体微阵列杂交(见图22-4下部,重复的红色信号)。与22q11.2微缺失不同的是,22q11.2微重复大多数来源于表型正常的父母,新发变异较为少见。
2q11.2微重复综合征患者表型呈高度异质性,症状的严重程度可以从无任何表型或轻微症状到严重畸形。22q11.2微重复综合征的表型通常轻微或不典型,故容易被漏诊,但该病患者有其特定的表型,主要表现为头面部畸形(上位眉毛、眼睑下斜或伴下垂、轻度的小颌/宿颌、长脸)、腭咽发育不全、腭裂、讲话鼻音重、先天性心脏畸形、听力障碍、肾生殖系统畸形、胸腺缺如、脾脏缺如、认知障碍等。
待续

 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国
