

蛋白质的功能很大程度上取决于其 3D 结构。19 世纪中期,科学界普遍认为蛋白质结构是固定的、刚性的,类似「锁与钥匙」模型 (lock-and-key model),即蛋白质与配体的结合是由固定的三维结构决定的。然而,当 Daniel Koshland 提出酶与底物结合时会发生构象变化的观点后,传统思维开始受到挑战。
1980 年代,分子动力学模拟 (Molecular Dynamics, MD) 兴起,首次从计算角度揭示了蛋白质的运动轨迹,自此,蛋白质动态结构的功能性作用受到越来越多的重视。对于生物技术研究人员和科学家而言,理解蛋白质「运动」的动态特征对理解生命过程、研发新型药物都具有重要的意义。
举例而言,G 蛋白偶联受体 (GPCR) 是许多药物的主要靶点,占目前 FDA 批准药物的 30% 以上。然而,GPCR 不是刚性结构,而是具有高度的动态性,不同的构象状态会影响药物结合模式。如果仅基于静态晶体结构设计药物,可能会错过关键的结合位点,导致药物亲和力和选择性不足。动态结构预测可以帮助识别 GPCR 在生理环境下的多种构象,从而优化小分子药物的设计,提高靶向治疗的成功率。
在此背景下,来自复旦大学、上海科学智能研究院的朱思语及漆远教授团队,联合南京大学姚遥教授,提出了一种创新性的 4D 扩散模型 AlphaFolding,结合分子动力学模拟数据,以学习动态蛋白质结构。这是首个基于扩散模型的方法,能够同时预测多个时间步长的蛋白质运动轨迹。
在基准数据集上的验证结果表明,该新模型在预测包含最多 256 个氨基酸、跨度达 32 个时间步长的动态 3D 结构方面表现出高精度,能够有效捕捉稳定状态下的局部柔性以及显著的构象变化。
相关成果以「4D Diffusion for Dynamic Protein Structure Prediction with Reference and Motion Guidance」为题,已入选国际顶会 AAAI 2025,预印本已发布于 arXiv。

蛋白质动态结构预测研究尚存空白
AlphaFolding 模型可以被视作结构生物学研究的重要进展。所谓结构生物学,是以蛋白质等生物大分子的结构、运动和相互作用的研究为基础来阐明生命现象的科学, 现已发展成为分子生物学的主流。
近年来,深度学习技术的进步,加上 Protein Data Bank (PDB) 实验蛋白质结构数据的指数级增长,使得蛋白质结构预测领域取得了不少重要突破。其中,最知名的莫过于 AlphaFold2,其使用最新的人工智能算法对蛋白质结构实现了接近实验精度的精准预测,相关成果被 Science 评为 2020 年十大科学突破之一。
无独有偶,2021 年 7 月,华盛顿大学生物学家 David Baker 团队研发的 RoseTTAFold 构建了一种「三轨 (three-track)」神经网络,能在十几分钟之内解析给定序列蛋白质的三维结构。
此外,大规模数据存储库的可用性促进了蛋白质构象采样研究的发展。例如,微软研究院开发出一种名为 Distributional Graphformer (DiG) 的深度学习框架,旨在预测分子结构在平衡态下的分布。传统的分子动力学模拟和增强采样方法虽然能够获得分子的平衡分布,但这些方法计算成本高且耗时,难以应用于复杂的实际应用场景。而 DiG 则是通过深度学习技术,能够快速生成真实多样的构象。
尽管蛋白质结构及其构象预测都取得了重大突破,但动态结构的研究仍然相对滞后。以 AlphaFold2 为例,其能够准确预测蛋白质的三维结构,但只能预测蛋白质在一个瞬间的静态结构,尚无法实现动态变化的预测。
2024 年 5 月,DeepMind 发布了升级后的 AlphaFold3,它能够以前所未有的「原子精度」预测出所有生物分子的结构和相互作用,包括蛋白质、核酸和更小分子的 3D 结构,并揭示它们如何组合在一起,但是其对生物分子的动态 3D 结构预测依然存在很大的局限性。
因此,本研究新提出的创新性 4D 扩散模型其实是为了弥补这一研究空白,重点关注蛋白质结构的动态特性,为更深入理解蛋白质功能提供新的思路。研究人员充分利用高质量的分子动力学模拟 (MD) 数据,生成包含完整侧链表示的动态蛋白质结构,适用于由数百个氨基酸组成的复杂蛋白质。这将扩展 MD 模拟的适用范围,使其能够预测更大、更复杂的蛋白质系统的动态行为,提升对蛋白质动态特性的理解。
在预测多个时间步长的蛋白质运动轨迹方面表现出高精度
静态蛋白质模型相对容易构建,那么动态的蛋白质模型该如何表示呢?为了解决这一问题,研究人员采用 AlphaFold2 的基于坐标框架 (frame-based) 蛋白质结构表示方法,并将其扩展至时间维度,以描述随时间变化的结构变换。
在静态蛋白质建模中,蛋白质由一系列氨基酸残基 (amino acid residues) 组成,每个残基由 backbone 框架进行参数化。而在本研究中,研究人员将动态蛋白质定义为包含 N 个氨基酸残基,且其 backbone 框架在 S 个时间步 (time steps) 内发生变换的系统。这些框架通过特殊欧几里得变换 (Special Euclidean Transformations) 进行变换,以保持局部框架到全局参考框架的方向。
蛋白质中的所有额外原子坐标根据与二面角 (torsion angles) 的依赖关系组织为刚性原子组 (rigid groups),以确保化学结构完整性。在每个刚性组内,所有原子相对位置和方向保持不变。结合变换参数 (transformation parameters),模型可以在时间维度上从理想化的实验坐标重建所有原子位置。
在此基础上,下图展示了整个研究模型构建的方法:该扩散模型以参考结构和对应的残基序列 (amino acid residues sequence) 作为输入,并生成一系列去噪后的 3D 蛋白质结构 (denoised 3D structure) 作为输出。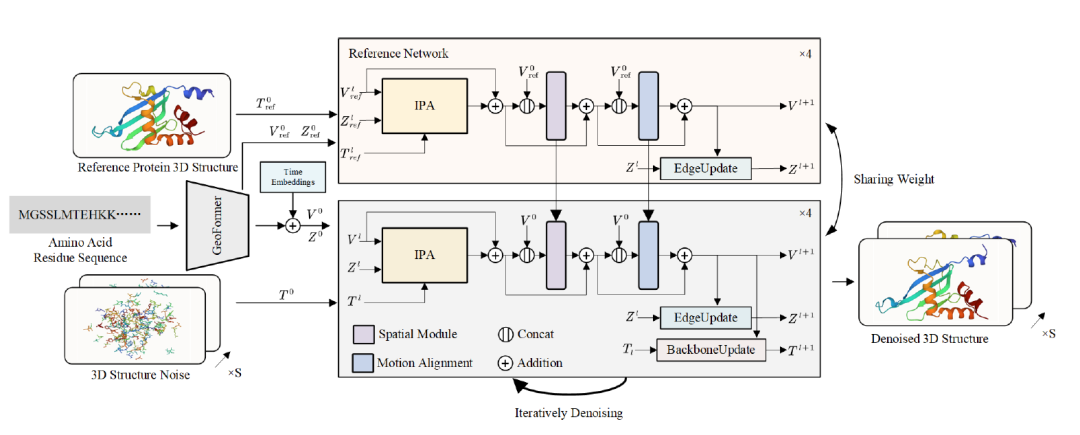
研究方法概述
研究人员使用 3D 结构嵌入器 (3D structure embedder) 和 GeoFormer 分别对 3D 蛋白质结构和残基序列进行嵌入。不变点注意力 (Invariant Point Attention, IPA) 通过结合残基的显式框架信息来更新节点特征。
参考网络 (Reference Network) 和运动对齐模块 (Motion Alignment module) 基于参考 3D 蛋白质结构捕捉 3D 蛋白质动力学序列。整个生成模型被构造为基于分数的扩散模型 (score-based diffusion model),其中节点和边的特征嵌入分别通过 EdgeUpdate 和 BackboneUpdate 模块进行更新。
构建模型后,研究人员将所提出的框架与当前短程到长程 (Short-term-to-long-term, S2L) 任务中的 DFF 和 Flow-Matching 进行了对比实验,所使用的数据集包括 ATLAS 和快速折叠蛋白 (Fast-Folding Proteins)。
结果如下表所示:在 ATLAS 数据集上的 S2L 任务,研究提出的方法 将 R32 误差从 4.60 降低至 2.12,显著提高了长期预测的准确性;在 Fast-Folding 数据集上的 S2L 任务,研究提出的方法将 R32 误差从 5.48 降低至 4.39,同样表现出良好的长期预测能力。同时,研究提出的模型在 O2O 任务上的表现与 S2L 任务相当,这表明其优秀的泛化能力。
DFF、FM 与本研究提出的方法在 ATLAS 蛋白质数据集上的 Cα-RMSE 比较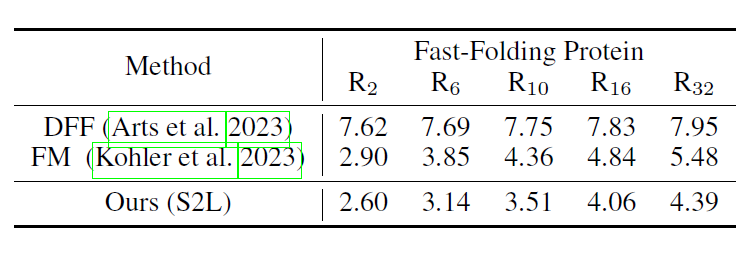
DFF、FM 与本研究提出的方法在 Fast-Folding 蛋白质数据集上的 Cα-RMSE 比较
此外,该方法能够处理包含较长模拟时间的蛋白质,这些蛋白在每个轨迹步长上的动力学变化更大。实验结果进一步验证了该方法在建模蛋白动力学方面的有效性 (efficacy in modeling protein kinetics)。
更进一步,研究人员还通过可视化模型生成前两个 TIC (时间一致性成分) 的动态蛋白质分布,并与真实数据进行比较。如下图所示,新模型有效地预测了蛋白质的动力学行为,与真实分布高度一致。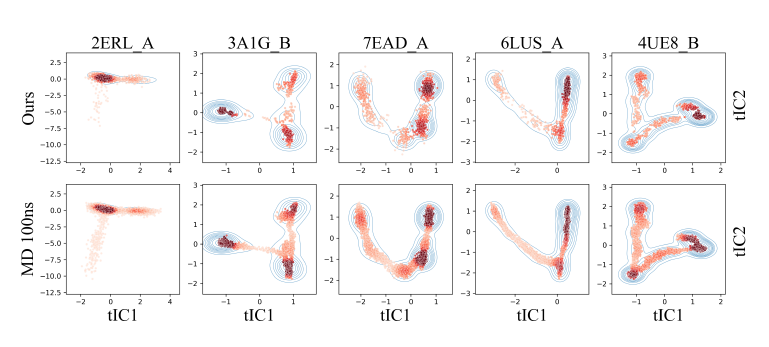
不同蛋白质在前两个 TIC 组件上的样本分布
* 点的颜色越深,出现频率越高,蓝色曲线表示从 MD 数据估计的核密度分布
下图则展示了在选定时间步上的逆扩散过程,突出了蛋白质结构在去噪过程中逐渐变得更加一致的过程。可以看到,所提出的方法有效地捕捉了蛋白质的动力学,生成了合理的轨迹。
从初始噪声 (左) 经过逆扩散过程逐步形成蛋白质结构 (右) 的可视化展示
* 粉色和黄色区域分别标注了 α 螺旋和 β 折叠
蛋白质结构的动态特性将受到更多关注
蛋白质在细胞环境中并非静态存在,而是处于复杂的动态变化之中。传统的静态结构预测方法虽然在揭示蛋白质折叠和相互作用方面取得了重要进展,但无法全面捕捉蛋白质的动态行为。因此,动态蛋白质结构预测成为结构生物学和计算生物学的前沿挑战之一,近年来,也有越来越多的研究人员投身于这一方向。
2022 年 12 月,西湖大学李子青团队与厦门大学、德睿智药合作,研发了能够刻画蛋白质构象变化与亲和力预测的 AI 模型 ProtMD。这是第一个尝试解析蛋白质动态构象的 AI 方法,给定药物分子和靶点蛋白,ProtMD 预测药物分子与生物体内靶点蛋白质结合后蛋白质结构的变化过程,推断药物与靶标蛋白结合的稳定性,预测药物功能,从而提升 AI 药物设计的精度和效率,加速临床前药物研发。
相关研究成果以「Pre-Training of Equivariant Graph Matching Networks with Conformation Flexibility for Drug Binding」为题,发表在 Advanced Science。
2024 年 8 月,康涅狄格大学的一项新研究揭示了一种先进的计算模型和工具,能够准确预测蛋白质的动态特征及其结晶倾向,相关研究成果以「Protein dynamics inform protein structure: An interdisciplinary investigation of protein crystallization propensity」为题发表在材料科学领域期刊 Matter 上。研究的重点在于蛋白质的自然运动和波动,即其摇摆特性,如何影响其功能属性,尤其是蛋白质形成高质量晶体的能力。
2024 年 10 月,上海交通大学郑双佳课题组联合星药科技、中山大学药学院以及美国莱斯大学,提出了为蛋白质动态对接 (dynamic docking) 设计的几何深度生成模型 DynamicBind,可以有效地将蛋白质构象从最初的 AlphaFold 预测状态调整到类似全息 (holo-like) 状态,为后 AlphaFold 时代的药物研发提供了一种基于深度学习的、考虑蛋白动态变化的新研究范式。
相关研究以「DynamicBind: predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model」为题,发表在 Nature Communications 上。
总而言之,动态蛋白质结构预测不仅能够帮助理解生命过程,还能在药物开发、疾病机理研究和工业生物技术等领域发挥重要作用。从 GPCR 药物设计、蛋白-蛋白相互作用,到酶催化和蛋白质聚集病理学研究,动态结构预测将不断推动生命科学的前沿发展。

 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国