
范围
GB 25572―20101本标准适用于以石灰石或含石灰石的牡蛎壳等为原料经煅烧、消化而成的食品添加剂氢氧化钙。
规范性引用文件本标准中引用的文件对于本标准的应用是必不可少的。凡是注日期的引用文件,仅所注日期的版本适用于本标准。凡是不注日期的引用文件,其最新版本(包括所有的修改单)适用于本标准。
分子式和相对分子质量分子式Ca(OH)2
相对分子质量74.09(按2007 年国际相对原子质量)
技术要求感官要求:应符合表1 的规定。
表1 感官要求
|| ||
理化指标:应符合表2 的规定。
表2 理化指标
|| ||
附录(规范性附录)检验方法
警示本标准检验方法中使用的部分试剂具有腐蚀性,操作时须小心谨慎!如溅到皮肤上应立即用水冲洗,严重者应立即治疗。使用剧毒品时,应严格按照有关规定管理;使用时应避免吸入或与皮肤接触,必要时应在通风橱中进行。暴露部位有伤口的人员不能接触。
一般规定本标准检验方法中所用试剂和水在没有注明其他要求时,均指分析纯试剂和GB/T 6682—2008 中规定的三级水。试验中所用标准滴定溶液、杂质标准溶液、制剂及制品,在没有注明其他要求时,均按HG/T3696.1、HG/T3696.2、HG/T3696.3 的规定制备。
鉴别试验试剂和材料
1 乙酸溶液:1+1。
2 草酸铵溶液:40g/L。
称取4g 草酸铵(C2H8N2O4·H2O)溶于100mL 水中。
3 红色石蕊试纸。
分析步骤
1 氢氧根离子的鉴别
称取约5g 样品,加入20mL 水混合,样品形成稠糊,稠糊上层的澄清液使红色石蕊试纸变蓝。
2 钙离子的鉴别
1g 样品与20mL 水混合,加足量乙酸溶液使样品溶解,加入草酸铵溶液,生成不溶的草酸盐沉淀。此沉淀不溶于乙酸而溶于盐酸。
氢氧化钙的测定方法提要
取适量试验溶液,加入蔗糖掩蔽碳酸盐的干扰,以酚酞为指示剂,用盐酸标准滴定溶液滴定至无色。
试剂和材料
1 盐酸标准滴定溶液:c(HCl)= 0.5mol/L。
2 蔗糖溶液:300g/L。
称取300g 蔗糖,溶于1000mL 水中。加1 滴酚酞指示液, 使用前滴加氢氧化钠溶液(4g/L)至溶液刚呈微粉色。
3 酚酞指示液:10g/L。
仪器和设备
电磁搅拌器。
分析步骤
称取约0.5g 的试样,精确至0.000 2g,置于250mL 锥形瓶中,加入50mL 水,振摇使之混匀。加入50mL 蔗糖溶液,用磁力搅拌器搅拌15min 后,加入2 滴~3 滴酚酞指示液,用盐酸标准滴定溶液滴定至试液刚变为无色,并保持30s 不返色即为终点。同时做空白试验,除不加试样外,其他加入的试剂量与试验溶液的完全相同(盐酸标准滴定溶除外),并与试样同时同样处理。
结果计算
氢氧化钙含量以氢氧化钙[Ca(OH)2]的质量分数w1 计,数值以%表示,按公式计算: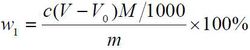
c——盐酸标准滴定溶液浓度的准确数值,单位为摩尔每升(mol/L);
V——试验溶液所消耗盐酸标准滴定溶液体积的数值,单位为毫升(mL);
V0——空白试验溶液消耗盐酸标准滴定溶液体积的数值,单位为毫升(mL);
m——试样的质量的数值,单位为克(g);
M——氢氧化钙[1/2Ca(OH)2]摩尔质量的数值,单位为克每摩尔(g/mol)(M=37.05)。
取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对差值不大于0.3%。
碳酸盐的测定试剂和材料
盐酸溶液:1+3。
分析步骤
称取约2g 试样,精确至0.01g,加入50mL 水,混匀,加入40mL 盐酸溶液,溶解过程应仅有细微的气泡生成。
镁及碱金属的测定试剂和材料
1 硫酸。
2 盐酸溶液:1+3。
3 氨水溶液:1+1。
4 草酸溶液:63g/L。
称取6.3g 草酸(H2C2O4·2H2O)溶解在100mL 水中。
5 甲基红指示液:1g/L。
仪器和设备
高温炉:温度可控制在800℃±25℃。
分析步骤
称取约0.5g 样品,精确至0.000 2g,加入10mL 水和6mL 盐酸溶液使试样溶解,煮沸1min。迅速加入40mL 草酸溶液,用力搅拌。加入2 滴甲基红指示液,滴加氨水溶液,至溶液呈黄色,冷却后将此混合液转移到100mL 容量瓶中,用水稀释至刻度,摇匀。静置4h 或过夜。用中速滤纸干过滤,弃去初始液10mL。用移液管移取50mL 滤液于已预先于800℃±25℃灼烧至质量恒定的瓷坩埚中,加入0.5mL 硫酸,水浴蒸发至近干(或于电炉上低温蒸发至近干)。再在电炉上细心蒸发至干。继续加热使铵盐完全分解并挥发。置于高温炉中,于800℃±25℃灼烧至质量恒定。
结果计算
镁及碱金属含量以质量分数w2 计,数值以%表示,按公式计算: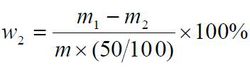
m1——瓷坩埚和残渣的质量的数值,单位为克(g);
m2——瓷坩埚的质量的数值,单位为克(g);
m—— 试料质量的数值,单位为克(g)。
取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对差值不大于0.2%。
酸不溶物的测定试剂和材料
1 盐酸溶液:1+3。
2 硝酸银溶液:17g/L。
仪器和设备
1 玻璃砂芯坩埚:孔径5μm~15μm。
2 电热恒温干燥箱:温度可控制在105℃±2℃。
分析步骤
称取约4g 试样,精确至0.0002g,加少量水润湿,加入60mL 盐酸溶液使试样溶解,加热煮沸。用预先于105℃±2℃干燥至质量恒定的玻璃砂芯坩埚趁热过滤中,用热水洗涤滤液至无氯离子(用硝酸银溶液检验)。置于电热恒温干燥箱中,于105℃±2℃干燥至质量恒定。置于干燥器中冷却至室温,称量。
结果计算
酸不溶物含量以质量分数w3 计,数值以%表示,按公式计算: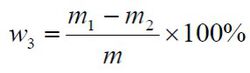
m1——玻璃砂芯坩埚和残渣的质量的数值,单位为克(g);
m2——玻璃砂芯坩埚的质量的数值,单位为克(g);
m—— 试料的质量的数值,单位为克(g)。
取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对差值不大于0.03% 。
砷的测定试剂和材料
1 盐酸溶液:1+3。
2 其它试剂同GB/T 5009.76—2003 的第9 章。
仪器和设备
同GB/T 5009.76—2003 的第10 章。
分析步骤
称取0.50g±0.01g 试样,置于锥形瓶中。加入10mL 盐酸溶液溶解试样。加4mL 盐酸,以下操作同GB/T 5009.76—2003 中第11 章“加水至30mL,再加5mL15%碘化钾溶液……不得深于砷的限量
标准的砷斑。”
限量标准溶液的配制:移取1.00mL 砷标准溶液(1mL 溶液含砷1.00μg),以下操作同GB/T 5009.76—2003 中第11 章“加5mL 盐酸……取出砷斑进行比较。”
氟化物的测定试剂和材料
1 盐酸溶液:1+11。
2 盐酸溶液:1+3。
3 乙酸钠溶液:3mol/L。
称取204g 乙酸钠(CH3COONa·3H2O),溶于300mL 水中,加乙酸溶液(1+16)调节pH 至7.0,加水稀释至500mL。
4 柠檬酸钠溶液:0.75mol/L。称取110g 柠檬酸钠(Na3C6H5O7·2H2O)溶于300mL 水中,加14mL 高氯酸,再加水稀释至500mL。
5 总离子强度缓冲剂。乙酸钠溶液与柠檬酸钠溶液等量混合,使用前配制。
6 氟化物标准溶液:1mL 溶液含氟(F)0.010mg。移取1.00mL 按HG/T 3696.2 配制的氟化物标准溶液,置于100mL 容量瓶中,用水稀释至刻度,摇匀。该溶液使用前配制。
仪器和设备
1 氟离子选择电极。
2 饱和甘汞电极。
3 电磁搅拌器。
4 电位计:分度值为0.02。
分析步骤
1 仪器的准备
将氟离子选择电极(按说明书活化)和饱和甘汞电极与测量仪器的负端与正端相连接,电极插入盛有水的50mL 塑料杯中,在电磁搅拌中(使用聚乙烯转子),读取平衡电位值,更换2 次~3 次水后,待电位值平衡后,即可进行电位测定。
2 测定
称取约1g 试样,精确至0.01g。置于50mL 烧杯中,加水润湿后,加入15mL 盐酸溶液使样品溶解。煮沸1min,冷却后,转移至50mL 容量瓶,加25mL 总离子强度缓冲剂,加水至刻度,摇匀。倒入50mL 塑料烧杯中测定电极电位。
3 工作曲线的绘制
分别移取1.00mL,2.00mL,4.00mL,5.00mL,6.00mL 氟化物标准溶液(相当于含氟0.010mg,0.020mg,0.040mg,0.050mg,0.060mg)于5 只50mL 容量瓶中,于各容量瓶中分别加25mL 总离子强度缓冲剂,10mL盐酸溶液,加水稀释至刻度,摇匀。倒入50mL 塑料烧杯中测定电极电位。以电极电位为纵坐标,氟的质量(mg)为横坐标,在半对数坐标纸上绘制工作曲线,根据试样的电位值在曲线上查得的氟的质量。
结果计算
氟化物含量以氟(F)的质量分数w4 计,数值以mg/kg 表示,按公式计算: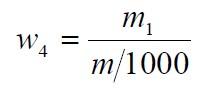
m1——根据测得的试验溶液电位值从工作曲线上查得的氟的质量的数值,单位为毫克(mg);
m ——试料的质量的数值,单位为克(g)。
取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对差值不大于5 mg/kg。
铅的测定石墨炉原子吸收分光光度法(仲裁法)
试剂和材料
1 硝酸。
2 硝酸溶液:0.5mol/L。将32mL 硝酸加入水中,稀释至1000mL。
3 磷酸二氢铵溶液:20g/L。称取2.0g 磷酸二氢铵,用水溶解,并稀释至100mL。
4 铅标准溶液:1mL 溶液含铅(Pb)100ng。
移取1.00mL 按HG/T 3696.2 配制的铅标准溶液置于100mL 容量瓶中,用硝酸溶液稀释至刻度,摇匀。再用移液管移取5mL 稀释过的溶液于500mL 容量瓶中,用硝酸溶液稀释至刻度,摇匀。该溶液使用前配制。
5 二级水:符合GB/T 6682—2008 的规定。
仪器和设备
1 所用玻璃仪器:均以硝酸溶液(1+5)浸泡过夜,用水反复冲洗,最后用去离子水冲洗干净。
2 原子吸收分光光度计(附石墨炉及铅空心阴极灯)。
分析步骤
1 试验溶液和空白试验溶液的制备
称取约0.5g 试样,精确至0.01g,置于50mL 烧杯中,用水润湿后,缓慢滴加约1.5mL 硝酸,低温加热,待样品完全溶解后,升温蒸至近干,取下,冷却至室温,补加2 滴硝酸,转移至25mL 容量瓶中,用水稀释至刻度,摇匀。同时配制空白试验溶液,此溶液除不加试样外,其他加入试剂的种类和数量与试验溶液相同,并同时操作。
2 工作曲线绘制
分别吸取铅标准溶液0.00mL,5.00mL,10.00mL,20.00mL,30.00mL,40.00mL 于6 个50mL 的容量瓶中,用硝酸溶液稀释至刻度,摇匀,此系列溶液浓度分别为0 ng/mL,10ng/mL,20ng/mL,40ng/mL,60ng/mL,80ng/mL。各吸取10μL,注入石墨炉,测得其吸光度,以铅的浓度为横坐标,对应的吸光度为纵坐标绘制工作曲线。
3 测定
在波长283.3nm 处将仪器调至最佳工作状态,分别吸取试验溶液和空白液10μL,注入石墨炉,测得其吸光度,从工作曲线上查得相应铅的浓度。
4 基体改进剂的使用
若有干扰,则注入适量的基体改进剂磷酸二氢铵溶液,一般为5μL 或与试样同量消除干扰。绘制铅标准曲线时也要加入与试样测定时等量的基体改进剂磷酸二氢铵溶液。
5 结果计算
铅含量以铅(Pb)的质量分数w5 计,数值以mg/kg 表示,按公式计算:
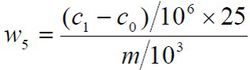
c1——从工作曲线上查出的试验溶液中铅的含量的数值,单位为纳克每毫升(ng/mL);
c0 ——从工作曲线上查出的空白试验溶液中铅的含量的数值,单位为纳克每毫升(ng/mL);
m ——试料的质量的数值,单位为克(g);
25——试料定容体积,单位为毫升(mL)。
取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对差值不大于1 mg/kg。
双硫腙分光光度法
试剂和材料
1 盐酸溶液:1+3。
2 其它试剂同GB/T 5009.75—2003 中第3 章。
仪器和设备
同GB/T 5009.75—2003 中第4 章。
分析步骤
称取约1g 试样,精确至 0.01g,置于50mL 烧杯中。加水润湿后,加入15mL 盐酸溶液使试样溶解,转移至125mL 分液漏斗中,加1%硝酸溶液至20mL。同时作空白试验,除不加试样外,其他加入的试剂量与试验溶液的完全相同,并与试样同时同样处理。以下同GB/T 5009.75—2003 中6.2“吸取铅标准溶液……绘制工作曲线”。
结果计算
铅含量以铅(Pb)的质量分数w6 计,数值以mg/kg 表示,按公式计算: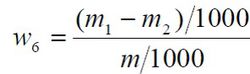
m1 ——从工作曲线上查出试验溶液中铅的质量的数值,单位为微克(μg);
m2 ——从工作曲线上查出空白试验溶液中铅的质量的数值,单位为微克(μg);
m ——试料的质量的数值,单位为克(g)。
取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对值不大于1 mg/kg。
重金属(以Pb计)的测定试剂和材料
1 盐酸溶液:1+1。
2 盐酸溶液:1+3。
3 氨水溶液:2+3。
4 乙酸盐缓冲溶液:pH﹦3.5。
称取25.0 g 乙酸铵,加25 mL 水溶解,加45 mL 盐酸溶液(A.11.1.1),再用盐酸溶液或氨水溶液调节pH 值至3.5,用水稀释至100 mL。
5 硫化钠溶液;
称取5 克硫化钠,加10mL 水溶解,加入30mL 丙三醇,混匀,加盖密封避光保存。配制后三个月内有效。
6 铅标准溶液:1mL 溶液含铅(Pb)0.01mg。
移取1.00mL 按HG/T 3696.2 配制的铅标准溶液,置于100mL 容量瓶中,用水稀释至刻度,摇匀。该溶液使用前配制。
7 酚酞指示液:10g/L。
仪器和设备
比色管:50mL。
分析步骤
称取2.00g±0.01g 试样,置于蒸发皿中,加水润湿后,加入 30mL 盐酸溶液使试样溶解,水浴蒸至干,加20mL 水使溶解,过滤于比色管中。加1 滴酚酞指示液,用氨水溶液调节至刚呈微粉色,加5 mL 乙酸盐缓冲溶液,用水稀释至刻度,加1 滴硫化钠溶液,摇匀,于暗处放置5 min。在白色背景下观察,所呈颜色不得深于标准比色溶液。
标准比色溶液的制备:移取2.00mL 铅标准溶液于比色管中,加水至20mL,加5 mL 乙酸盐缓冲溶液,用水稀释至刻度,加1 滴硫化钠溶液,摇匀,于暗处放置5 min。与试料同时处理。
干燥减量的测定仪器和设备
1 称量瓶:Φ40×25mm。
2 电热恒温干燥箱:温度可控制在105℃±2℃。
分析步骤
称取约2g 试样,精确至0.0002g,置于预先于105℃±2℃下干燥至质量恒定的称量瓶中,置于电热恒温干燥箱,在105℃±2℃下干燥1h。取出,于干燥器中冷却至室温,称量。
结果计算
干燥减量以质量分数w7 计,数值以%表示,按公式计算: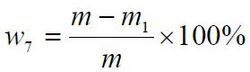
式中:
m——干燥前试料的质量的数值,单位为克(g);
m1 ——干燥后试料的质量的数值,单位为克(g);
取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对值不大于0.03%。
筛余物的测定仪器和设备
1 试验筛:R20/3 系列,Ф200×50—0.045/0.032 GB/T 6003.1 —1997。
2 软毛刷。
分析步骤
称取约10g 试样,精确至0.01g。移入试验筛内,用软毛刷轻刷试样,使粉末通过,最后,在筛子下垫一张黑纸,轻刷筛子直至所垫黑纸上没有试样痕迹。将筛余物转移到已知质量的表面皿中称量,精确至0.0002g。
结果计算
筛余物含量以质量分数w8 计,数值以%表示,按公式计算:
m1——筛余物的质量的数值,单位为克(g);
m ——试料的质量的数值,单位为克(g)。
取平行测定结果的算术平均值为测定结果,两次平行测定结果的绝对差值不大于0.04%。
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国